
SAN DIEGO – For thirty thousand Americans carrying a genetic defect in a single gene, Huntington’s disease has been a one-way street. The mutation accumulates silently through childhood and early adulthood, then starts dismantling movement, memory, and personality with a precision that no approved therapy has yet been able to interrupt.
A study published Wednesday in Nature offers an early indication that one key part of that dismantling process can, at least in mice, be reversed. Researchers at the University of California San Diego, working with collaborators at the Max Planck Institute for Biological Intelligence in Germany, identified a population of brain cells whose activity collapses early in Huntington’s disease and demonstrated that restoring those cells’ function improved motor learning in affected mice for multiple days after stimulation ended.
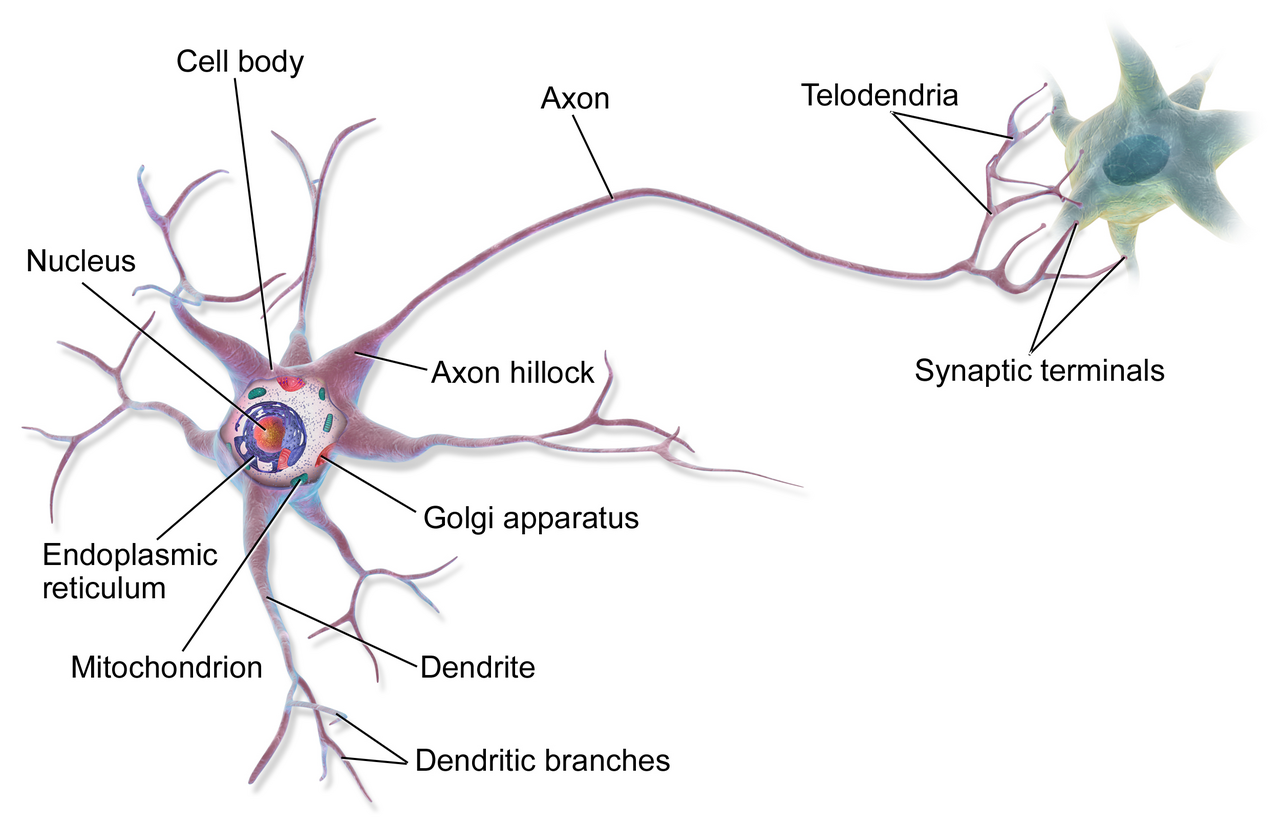
The cells in question belong to a class of inhibitory neurons in the motor cortex known as vasoactive intestinal peptide neurons, or VIP neurons. The finding is surprising in part because Huntington’s research has historically focused on excitatory cells, the neurons whose disrupted firing produces the involuntary movements and coordination failures that define the disease’s outward clinical picture. “Cortical inhibitory cells have received little attention in Huntington’s disease, as for a long time they were considered to be spared from neurodegeneration,” said Irina Dudanova, who helped lead the study and now directs a research group at the University of Würzburg. The imaging data produced by her team and UC San Diego neuroscientist Takaki Komiyama showed something different: VIP neurons were substantially less active in affected animals, disrupting the motor cortex’s ability to regulate its own signals.
VIP neurons play a specific architectural role in the brain’s movement circuitry. They are inhibitory neurons that suppress other inhibitory neurons, a configuration neuroscientists call disinhibition. When VIP neurons are active, they selectively silence competing inhibitory signals and allow specific movement-related activity patterns to emerge more cleanly. In the Huntington’s mice, this gating function was degraded, and the team’s imaging showed the consequent disruption cascading through excitatory cell populations that had previously seemed to be the primary site of the disease’s damage.
To test whether the VIP neuron deficit was causally connected to the motor learning impairments seen in Huntington’s mice, the researchers turned to optogenetics, a technique that uses light delivered through fiber optics to activate specific neurons with millisecond precision via a genetically introduced photosensitive protein. By targeting VIP cells during trained movement tasks, they could restore the disinhibition signal the disease had suppressed.
The results were measurable and more durable than the team anticipated. “By activating the VIP inhibitory cell type, we gradually restored more normal activity patterns, and, very importantly, we also saw an improvement in the ability of the mouse to learn a motor task,” said Sonja Blumenstock, an assistant project scientist at UC San Diego and one of the paper’s lead authors. That alone would have been a significant finding. The improvement did not vanish when the stimulation stopped. “The improvements persisted for days after stimulation ended,” said Takaki Komiyama, a professor in UC San Diego’s departments of neurobiology and neurosciences who led the effort, “suggesting that the treatment triggered lasting beneficial changes in brain circuits rather than only temporary effects.”
That persistence implies something more durable than a signal patch. It suggests that the circuits themselves, given a period of corrected input, can partially re-establish patterns that the disease had been degrading. Whether that interpretation holds under more rigorous follow-up experiments remains to be seen, but it matters to any future application because non-invasive stimulation techniques, which are the only realistic path to human treatment, would almost certainly need to be intermittent. Sustained benefits from episodic intervention would make such approaches far more plausible.
Optogenetics cannot be applied to humans in its current form. It requires genetic modification of neurons to introduce the light-sensitive protein, combined with surgically implanted hardware to deliver the light signal. It is a research tool that opens questions rather than answers them. What the UC San Diego results do establish is a specific cellular target and a mechanism through which that target can be modulated. “If we know which cells to target,” Dudanova said, “we can retune the brain’s abnormal activity patterns. This gives hope for future therapies.” Non-invasive brain stimulation methods already in clinical use for conditions including depression and Parkinson’s disease could potentially be refined to reach VIP neuron populations without genetic modification, though no such technique yet exists for this specific application.
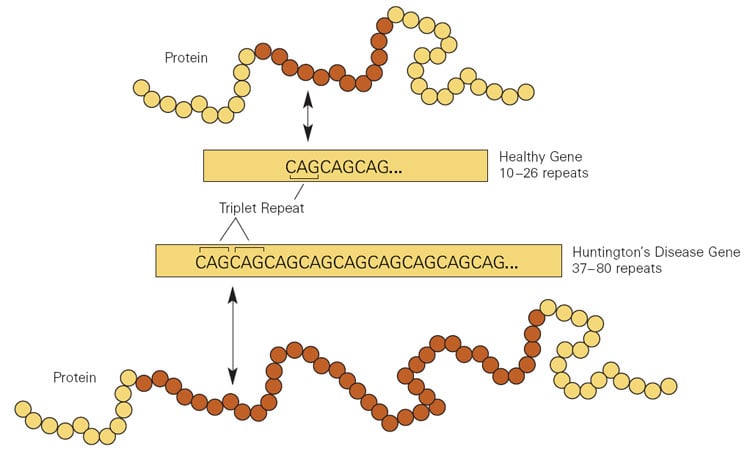
Huntington’s disease is caused by an abnormal expansion of a CAG trinucleotide repeat in the HTT gene. More than roughly 36 repetitions cause the mutant huntingtin protein to become toxic to neurons, progressively damaging the striatum and cortex over decades. The condition affects an estimated 30,000 Americans, with roughly 200,000 more carrying the mutation without yet showing symptoms. Every child of an affected parent has a 50 percent chance of inheriting the mutation, and there is no approved disease-modifying treatment. Current therapies manage symptoms without altering the disease’s course.
The new research fits within a broader scientific reassessment of when, precisely, intervention in neurodegenerative disease is most likely to work. A McMaster University team publishing in the same journal last week made a related argument about glioblastoma: that targeting the disease’s early influence on its microenvironment, rather than waiting for the final cell-killing stage, is where effective interventions may actually exist. The Huntington’s findings suggest something comparable. Circuit-level disruptions appear before widespread neurodegeneration becomes irreversible, and may be where the disease creates its most correctable damage.
The research was funded by the National Institutes of Health, the National Science Foundation, the Chan-Zuckerberg Initiative, the Max Planck Society, Germany’s Deutsche Forschungsgemeinschaft, the European Research Council, and the Simons Collaboration on the Global Brain. The breadth of support, spanning three countries and several of the largest private scientific foundations in neuroscience, reflects both the severity of the disease and the distance that still separates a mouse experiment from a clinical treatment.
The paper, published July 1 in Nature, carries a finding that opens questions the authors themselves say they cannot yet answer. The transgenic mice in these experiments carry the same genetic mutation that causes human Huntington’s disease. But they do not experience the disease as a human patient does, over four decades of progressive motor, cognitive, and psychiatric deterioration across brain regions that no mouse lives long enough to fully replicate. A circuit that can be retrained in a mouse over days of stimulation may behave very differently inside a brain that has been accumulating damage for twenty years. The researchers do not claim otherwise. What they found is a cellular mechanism worth targeting. Whether targeting it leads to anything useful for people with Huntington’s disease, and how long finding out will take, remains genuinely unknown.